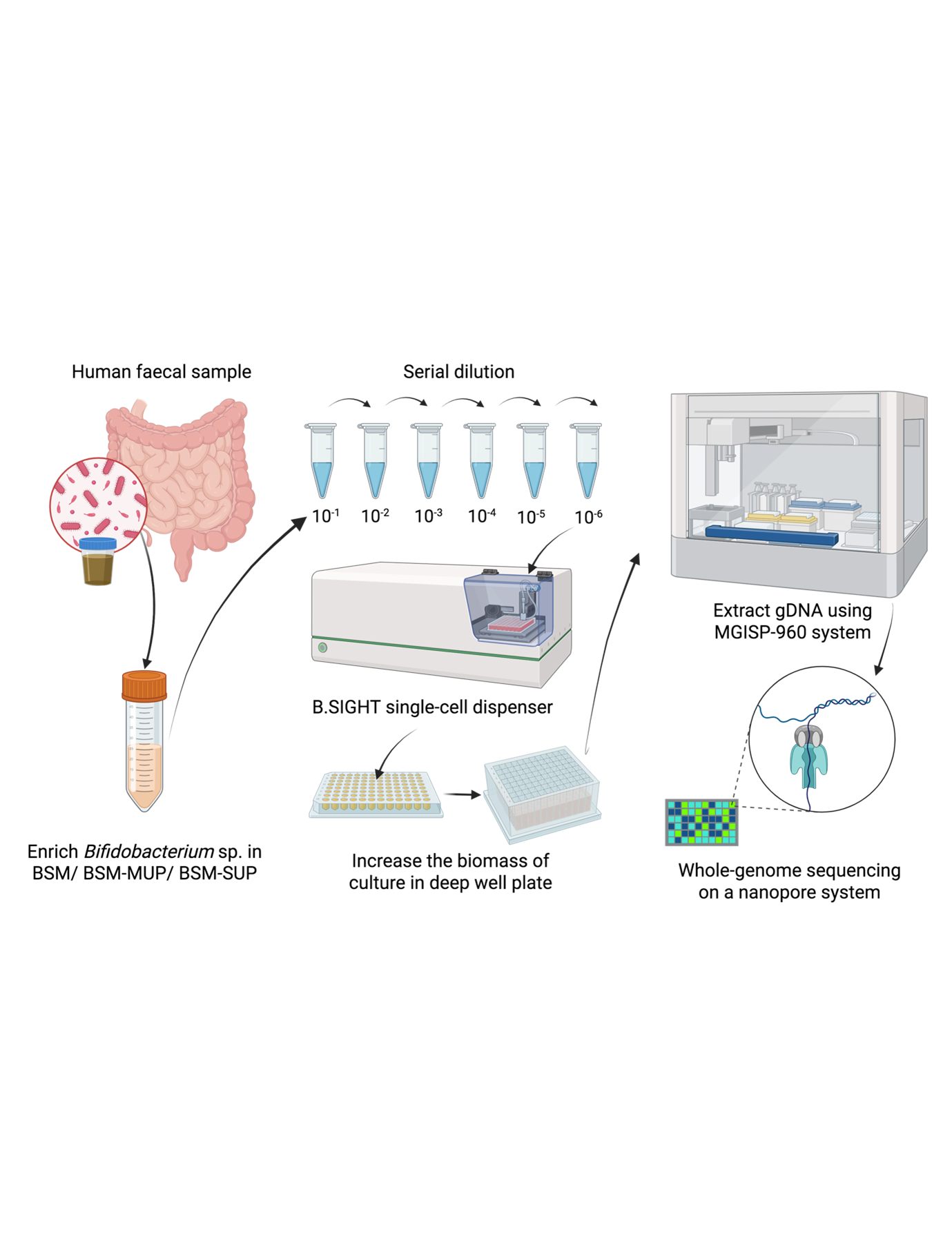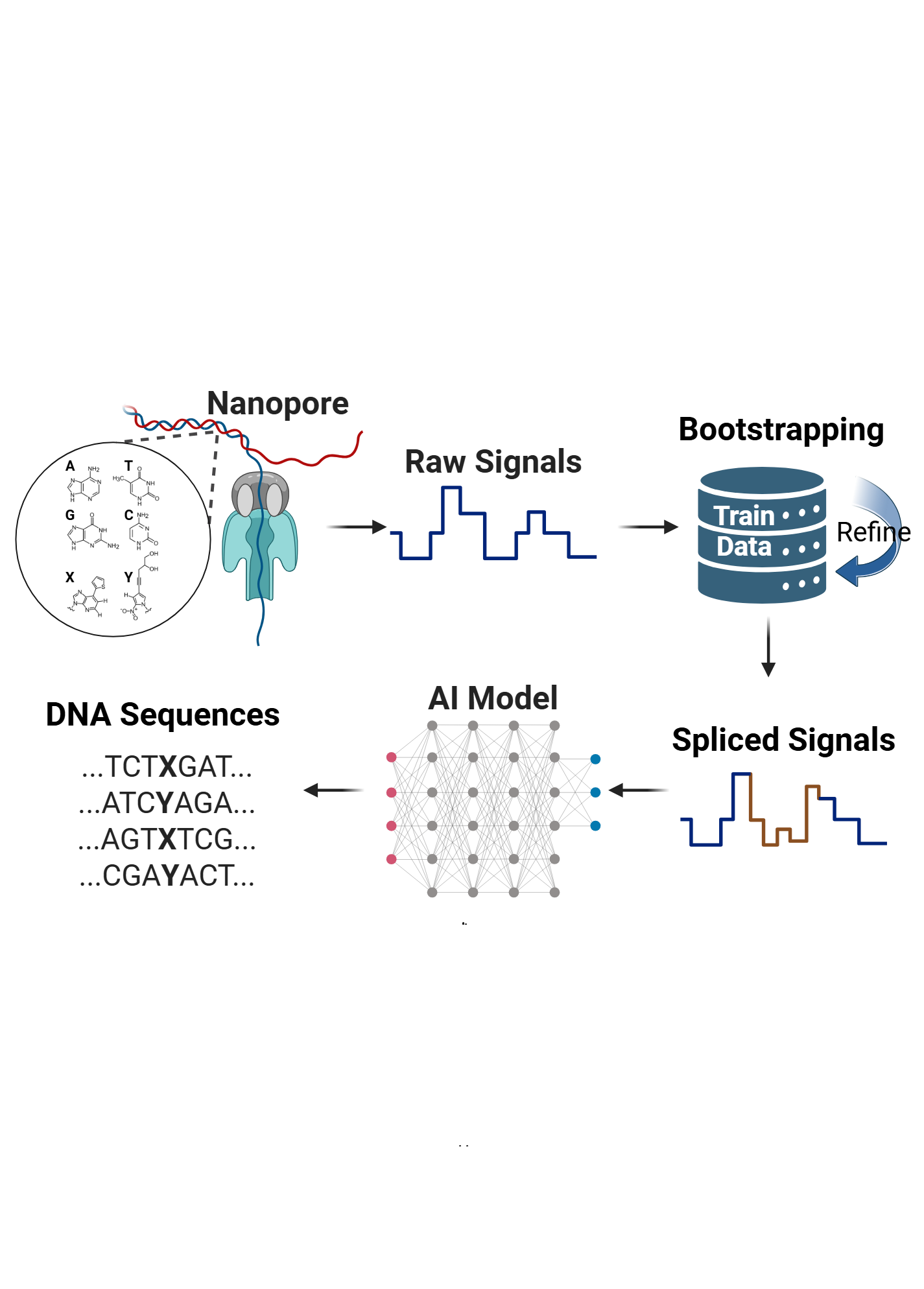


Given a set of sequenced reads, how can we determine if the sequencing run is good or not? Finding the quality of the reads, which includes estimating sequencing error rates and bias, has been an important first step in numerous Bioinformatics pipelines.
Previous ways of estimating sequencing error rates include mapping the reads to reference genomes and inferring error rates from Phred quality scores. Reference genomes may however be missing or different from the genomes that are actually sequenced, especially in metagenomic samples. On the other hand, Phred quality scores can produce biased estimates if they are uncalibrated.
We therefore propose a new framework for estimating sequencing error and bias, called skiver, which works without the need for reference genome or relying on Phred scores.
 Workflow of skiver.
Workflow of skiver.
The key ideas in skiver is to use (k, v)-mer sketches to represent the large amount of sequencing reads. A (k, v)-mer is a segment of length k+v, where the first k bases are the key and the last v bases are the value. By grouping the (k, v)-mers with the same key together, we can identify the consensus value, as well as estimate the frequency of sequencing errors.
Experiments on various real datasets show that skiver is able to accurately estimate the sequencing error rate and infer the percentage of k-mers in the read set that are free of sequencing errors. In addition, skiver can estimate the substitution, insertion, and deletion rates, revealing the biases of various sequencing platforms.
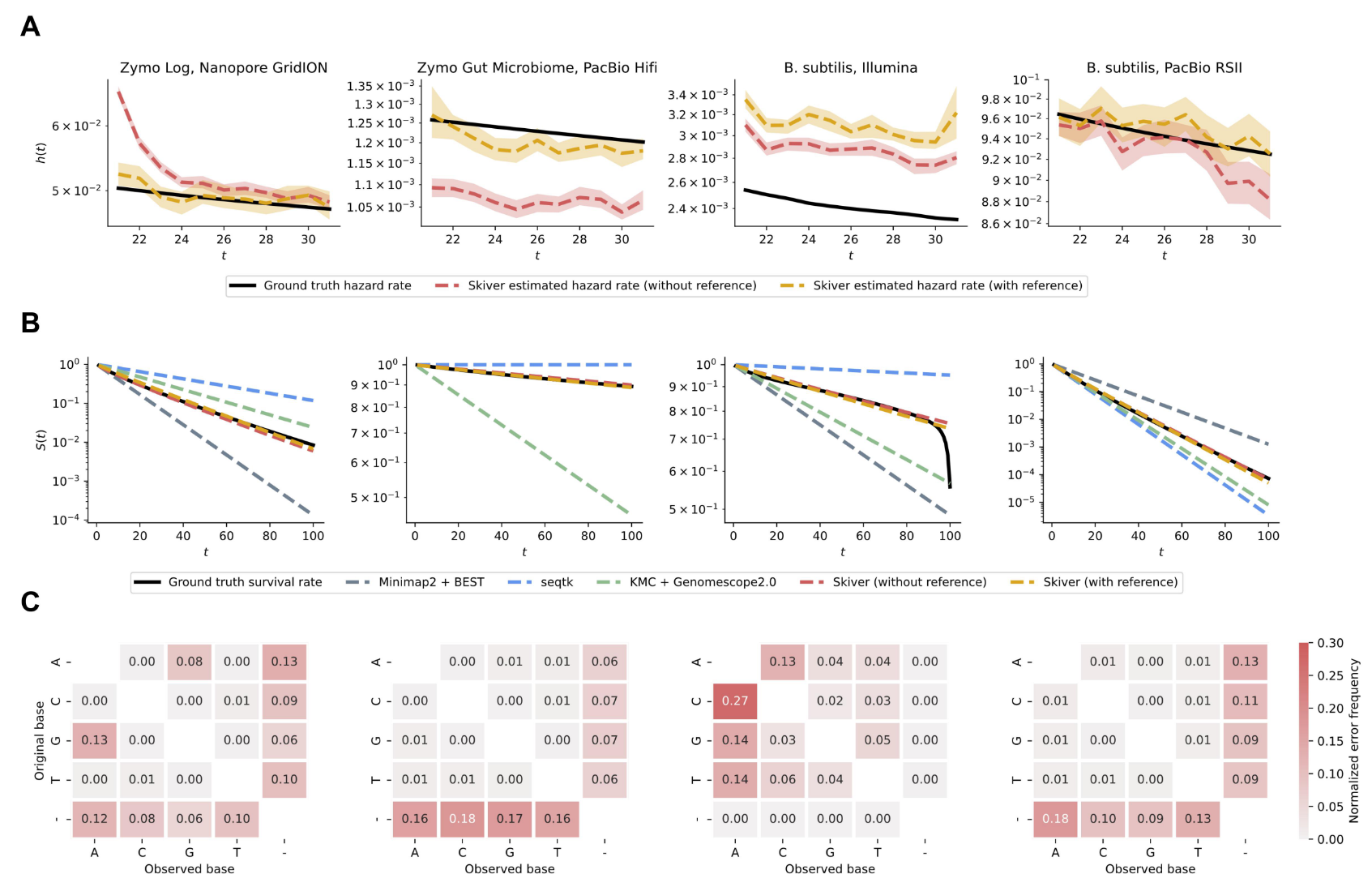 Skiver’s estimation of error rates and error spectra on various metagenomic samples.
Skiver’s estimation of error rates and error spectra on various metagenomic samples.
Finally, skiver is computationally lightweight, making it a handy tool for quality control in modern Bioinformatic pipelines.
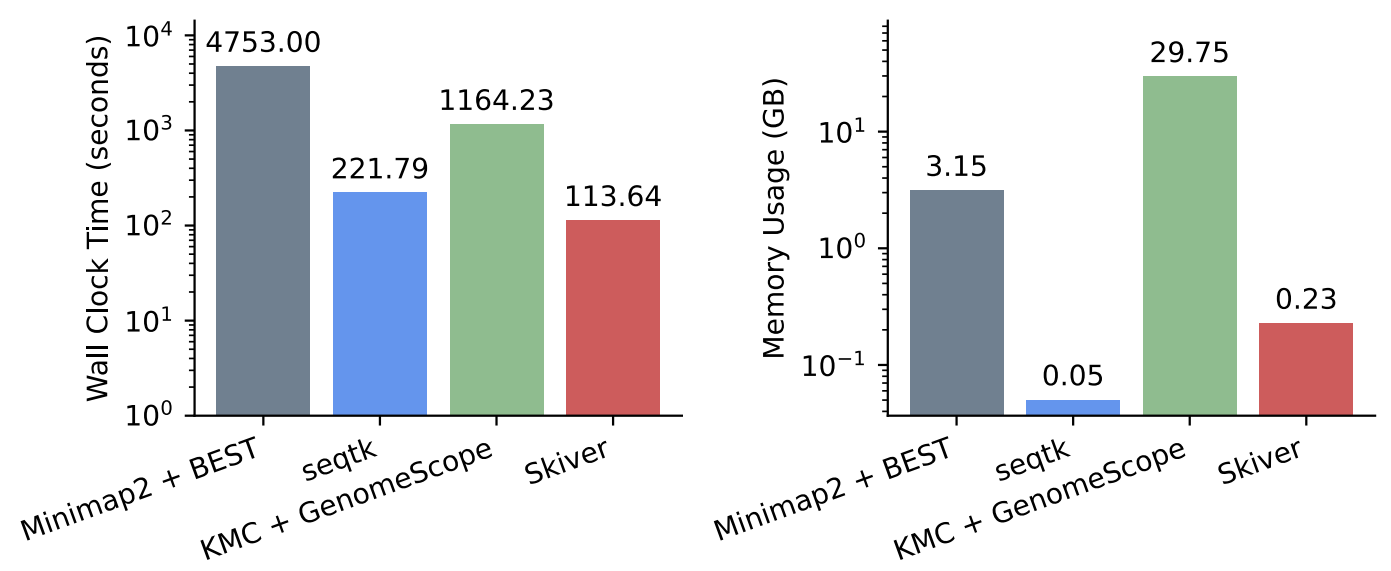 Computational resources needed by skiver and other baselines.
Computational resources needed by skiver and other baselines.